RNA distance distribution to nuclear envelope¶
Workflow to quantify the distance distribution of RNAs to the cell membrane.
- FISH-quant to detect RNA positions.
- ImJoy: you can install the plugin from here.
ImJoy plugins will be available in the workspace: rna-loc-elegans
You will also need the ImJoy plugin engine, please consult the ImJoy documentation
Analysis overview¶
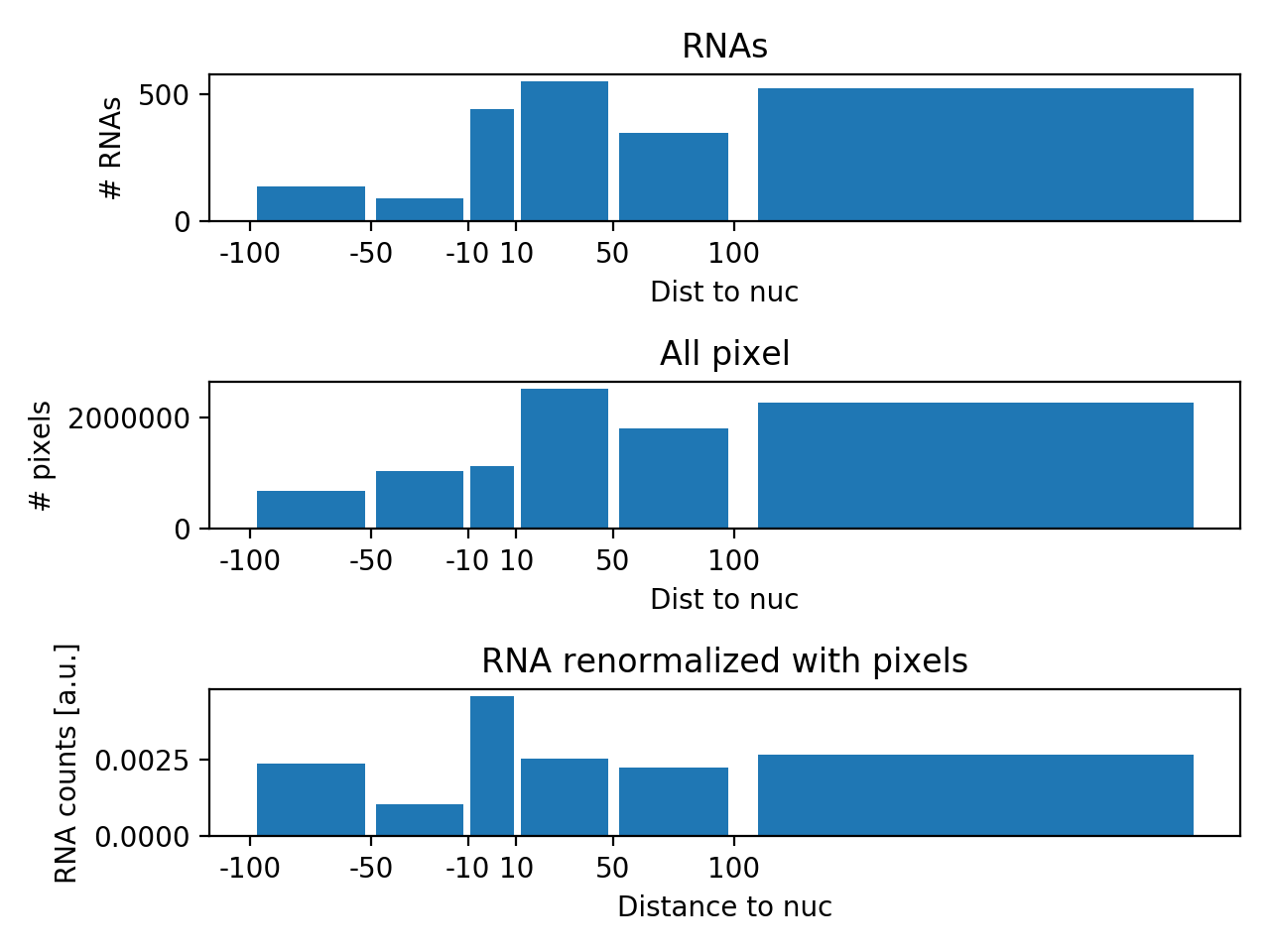
For each RNA, we determine the closest distance of an RNA to the nuclear envelope. This distance is negative for an RNA inside a nucleus, and positive for an RNA outside of a nucleus. Distances for all RNAs will be summarised in histogram. These counts are then normalised as follows:
- Normalisation for complete spatial randomness. There is "more space" away from a nucleus than close to it. This means that for a randomly distributed RNA it is less likely to be close to a nucleus, then being further away. We consider this by calculating a histogram with the distance of all pixels in the embryo to the nuclei. The RNA counts are then normalised with this pixel histogram.
- The normalised histogram is then further normalised such that it sums up to 1.
Data organisation¶
We enforce a strict data organisation for this analysis. More information about can be found in the dedicated section below.
- One folder per multi-channel image ('sample folder').
- Each channel is saved as a separate
.tiffile containing all z-slices. Information for how to convert stacks can be found here are here. - RNA molecules are detected with FISH-quant. Analysis results are also stored directly in the sample folder. RNA detection is described in more detail here. When performing RNA detection with
the GMM approach, the GMM results (ending with
_res_GMM.txt) has to be copied in the sample folder. - Annotations of nuclear envelope are stored in a dedicated subfolder
zstack_segmentation. More information below. - The outline of the embryo has to be store as a single FIJI ROI named
embryo_contour.roi.
As an example, we have one image img1 with 3 channels. The first channels contain
analysed smFISH data, the third one contains the cell membrane annotations for two Z slices (slice 3 and 8).
.
├─ img1/
│ ├─ C1-img1.tif # smFISH image (channel 1)
│ ├─ C1-img1__spots.txt # FQ detection results (channel 1)
│ ├─ C1-img1__settings_MATURE.txt # FQ detection settings (channel 1)
│ ├─ C2-img2.tif # smFISH image of (channel 2)
│ ├─ C2-img2__spots.txt # FQ detection results (channel 2)
│ ├─ C2-img2__settings_MATURE.txt # FQ detection settings (channel 2)
│ ├─ embryo_contour.roi # Embryo outline
│ ├─ zstack_segmentation/
│ │ ├─ C3-img1_Z3.tif # Image of z-slice 1
│ │ ├─ C3-img1_Z3__RoiSet.zip # Membrane annotation of slice 1
│ │ ├─ C3-img1_Z8.tif # Image of z-slice 2
│ │ ├─ C3-img1_Z8__RoiSet.zip # Membrane annotation of slice 2
├─ img2/
.
Test data¶
You can download already processed test data for the Cell membrane enrichment plugin, from the GitHub release page.
The zip archive contains data following the naming conventions of the examples below.
Annotations of nuclear envelope¶
In order to be able to compute the enrichment towards cell membrane or nuclear envelope, these membranes have to be annotated in EACH z-slice that should be considered in the analysis. This annotation can either be done manually, or automatically. If an automated segmentation is performed, make sure that the results are stored as described below for the manual annotation.
Manual with FIJI¶
Annotations have to be performed and stored as described below.
- For each multi-channel image, the annotations are stored in a dedicated sub-folder
called
zstack_segmentation. -
Each annotated z-slice has to be saved as an individual image and the file name has to end with the corresponding z-slice
iin the format_Zi. For instance, file name ends withimg1_ch3_Z3.tif, where3is the actual slice number.You can obtain the individual slices with different methods:
- FIJI. Open the z-stack and go to menu
Image>Stack>Stack to images. You can then save each image with the according file-name.
- FIJI. Open the z-stack and go to menu
-
Annotations are stored with the name of the corresponding image, with the additional suffix
__RoiSet.zip. For the example above, the added suffix would beimg1_ch3_Z3__RoiSet.zip.
To perform the actual annotation, follow these steps
- Open FIJI.
- Open the ROI manager:
Analyze>Tools>ROI manager - Open/Select the image that you would like to annotate.
- Select the annotation tool of choice, e.g. freehand or polygon.
- Outline first structure of interest. When done, press
Add(t)in the ROI manager to add outline to the out line manager. - Proceed with all other structures. Enabling
Show all, will show all defined regions. - Save regions by highlighting all regions in the list and pressing on
More>>Save ... - If only one region is saved, this will created a file with the extension
.roi, if multiple regions will be save this will create a.zipfile. As a file-name choose the name of the annotated image, followed by the suffix**__RoiSet.zip**.
IMPORTANT: these annotations could be used for training of a neural network. If this is intended, then ALL structures, e.g. nuclei, have to be annotated. Unwanted elements, e.g. nuclei touching the cell border, can be removed in a post-processing step.
Analysis with ImJoy¶
The entire functionality of the plugin can be controlled within the plugin window. If you expand the plugin, you will see all parameters of the plugins.

Analysis parameters¶
If you change these parameters, the changed values will be saved in the browser and reused the next time you launch ImJoy.
| Option | Type | Default | Description |
|---|---|---|---|
dZ |
int | 0 | Number of z-slices that will be used above and below the currently analysed slice. Specify this value > 0 if not all z-slices are annotated/ |
Zmin |
int | 0 | Specify at which Z slice analysis should start. Value of 0 means that entire z-stack will be processed. |
Zmax |
int | 0 | As Z min but for maximum Z slice. |
Hist [bins] |
string | ... | Defines the edges of the bins that will be used. Values are separated by a comma. |
FQ file |
str | FULL path name for FQ file to be analyzed. |
Launch analysis and monitor progress¶
To analyse a FQ results file, you have to
- Specify the required parameters in the plugin interface. This includes to copy & paste the FULL name of the FQ results file that should be analyzed. This can either be a 'regular' FQ detection, or an analysis with a GMM.
- Execute plugin with clicking on the Plugin name
NuclearEnvelopeEnrichment. - This file will then be processed and the final distance enrichment histogram displayed in the interface. More plots and result files will be stored as detailed below.
Once you start the analysis, ImJoy will show you a progress window with progress-bars for the different steps. Once done, it will display the final result in this image.
Stored results¶
The analysis script will create a new folder with the same name as the selected results file. To allow to redo the analysis with different settings, the plugin will create for each performed analysis a separate sub folder NucEnvelopDist_yymmdd-hhmm, where is a time-stamp in the format yymmdd-hhmm. This folder contains several files:
-
_DistanceEnrichmentSummary.png: image containing the pooled analysis of all slices.
-
_HistogramDistances.csv: csv file containing the histograms of the RNA-membrane distance pooled from all z-slices. This can be opened with any Spreadsheet application (Excel, LibreOffice, Numbers, ...) and results from different images can then be pooled together. It contains the following data
center: center of the binswidth: width of the binshistRNA_all: raw RNA distance histogram.histPIX_all: normalized RNA histogram (values add to 1).histRNA_norm: normalized RNA histogram with the pixel histogram.
-
DataAll.json: json file containing all analysis results including settings.